Note
Go to the end to download the full example code
Visualizing Single-cell RNA-seq Data#
import matplotlib as mpl
import matplotlib.pyplot as plt
from matplotlib.colors import Normalize
import marsilea as ma
import marsilea.plotter as mp
from sklearn.preprocessing import normalize
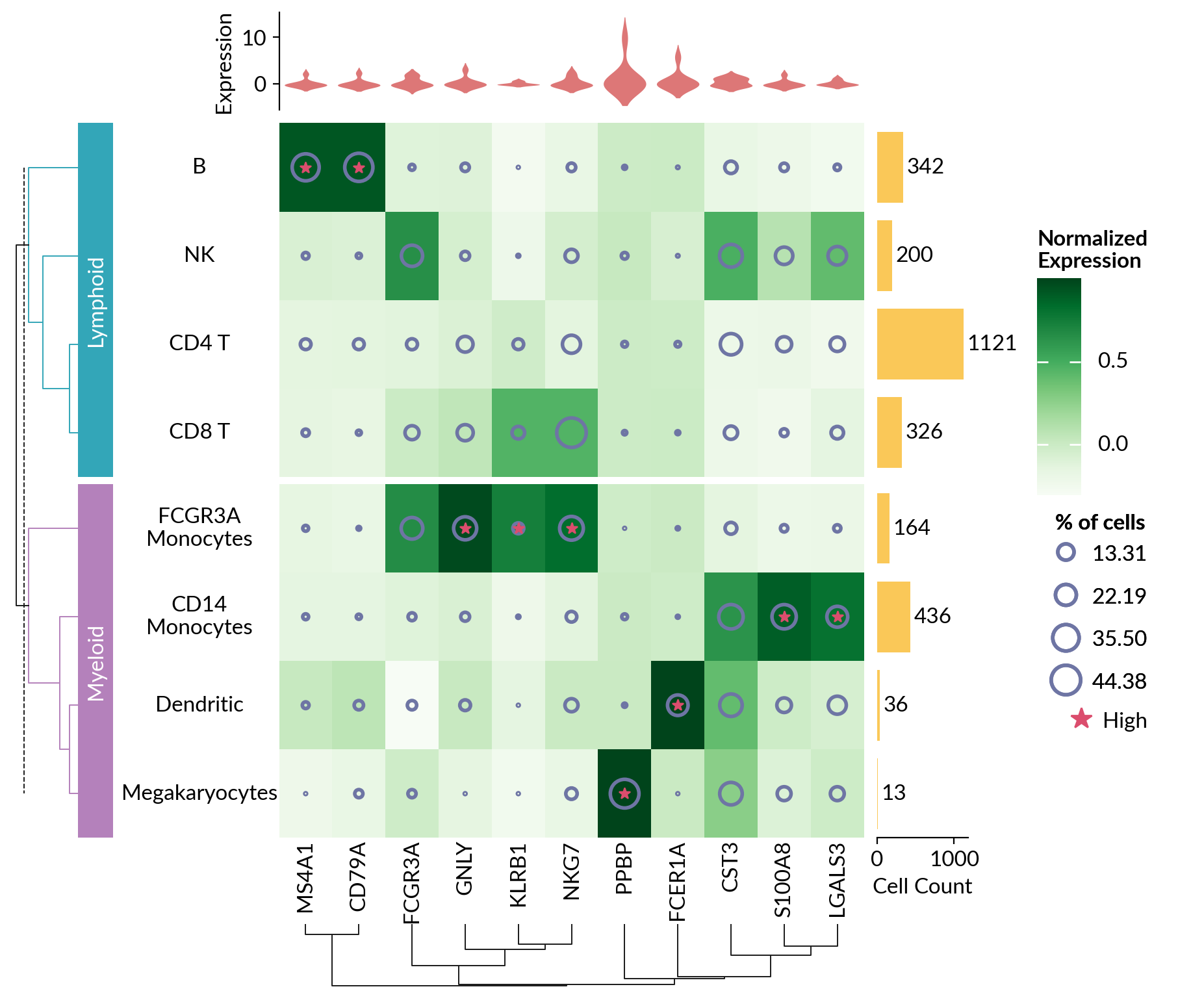
pbmc3k = ma.load_data("pbmc3k")
exp = pbmc3k['exp']
pct_cells = pbmc3k['pct_cells']
count = pbmc3k['count']
matrix = normalize(exp.to_numpy(), axis=0)
cell_cat = ['Lymphoid', 'Myeloid', 'Lymphoid', 'Lymphoid',
'Lymphoid', 'Myeloid', 'Myeloid', 'Myeloid']
cell_names = ['CD4 T', 'CD14\nMonocytes', 'B', 'CD8 T', 'NK',
'FCGR3A\nMonocytes', 'Dendritic', 'Megakaryocytes']
# Make plots
cells_proportion = mp.SizedMesh(
pct_cells, size_norm=Normalize(vmin=0, vmax=100),
color="none", edgecolor="#6E75A4", linewidth=2, sizes=(1, 600),
size_legend_kws=dict(title="% of cells", show_at=[.3, .5, .8, 1])
)
mark_high = mp.MarkerMesh(matrix > 0.7, color="#DB4D6D", label="High")
cell_count = mp.Numbers(count['Value'], color="#fac858", label="Cell Count")
cell_exp = mp.Violin(exp, label="Expression", linewidth=0, color="#ee6666",
density_norm="count")
cell_types = mp.Labels(cell_names, align="center")
gene_names = mp.Labels(exp.columns)
# Group plots together
h = ma.Heatmap(matrix, cmap="Greens", label="Normalized\nExpression",
width=4.5, height=5.5)
h.add_layer(cells_proportion)
h.add_layer(mark_high)
h.add_right(cell_count, pad=.1, size=.7)
h.add_top(cell_exp, pad=.1, size=.75, name="exp")
h.add_left(cell_types)
h.add_bottom(gene_names)
h.hsplit(labels=cell_cat, order=['Lymphoid', 'Myeloid'])
h.add_left(mp.Chunk(['Lymphoid', 'Myeloid'], ["#33A6B8", "#B481BB"]),
pad=.05)
h.add_dendrogram("left", colors=["#33A6B8", "#B481BB"])
h.add_dendrogram("bottom")
h.add_legends("right", align_stacks="center", align_legends="top", pad=.2)
h.set_margin(.2)
h.render()
# h.get_ax("exp").set_yscale("symlog")
Total running time of the script: (0 minutes 2.617 seconds)