Note
Go to the end to download the full example code.
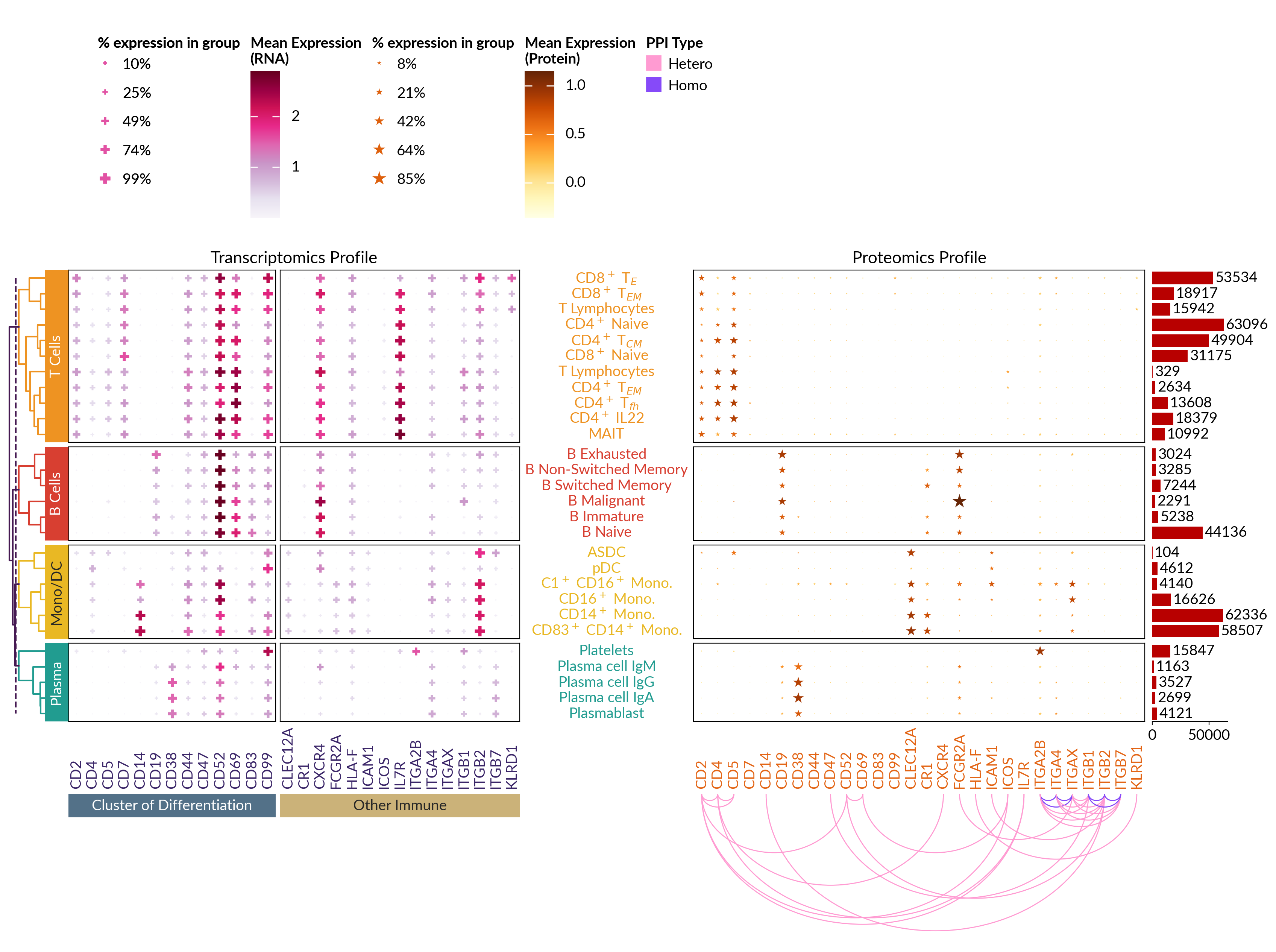
Visualizing Single-cell Multi-Omics Data#
import matplotlib as mpl
from matplotlib.ticker import FuncFormatter
import marsilea as ma
dataset = ma.load_data("sc_multiomics")
fmt = FuncFormatter(lambda x, _: f"{x:.0%}")
lineage = ["B Cells", "T Cells", "Mono/DC", "Plasma"]
lineage_colors = ["#D83F31", "#EE9322", "#E9B824", "#219C90"]
m = dict(zip(lineage, lineage_colors))
cluster_data = dataset["gene_exp_matrix"]
interaction = dataset["interaction"]
lineage_cells = dataset["lineage_cells"]
marker_names = dataset["marker_names"]
cells_count = dataset["cells_count"]
display_cells = dataset["display_cells"]
with mpl.rc_context({"font.size": 14}):
gene_profile = ma.SizedHeatmap(
dataset["gene_pct_matrix"],
color=dataset["gene_exp_matrix"],
height=6,
width=6,
cluster_data=cluster_data,
marker="P",
cmap="PuRd",
sizes=(1, 100),
color_legend_kws={"title": "Mean Expression\n(RNA)"},
size_legend_kws={
"colors": "#e252a4",
"fmt": fmt,
"title": "% expression in group",
},
)
gene_profile.group_rows(lineage_cells, order=lineage)
gene_profile.add_left(ma.plotter.Chunk(lineage, lineage_colors, padding=10))
gene_profile.add_dendrogram(
"left",
method="ward",
colors=lineage_colors,
meta_color="#451952",
linewidth=1.5,
)
gene_profile.add_bottom(
ma.plotter.Labels(marker_names, color="#392467", align="bottom", padding=10)
)
gene_profile.cut_cols([13])
gene_profile.add_bottom(
ma.plotter.Chunk(
["Cluster of Differentiation", "Other Immune"],
["#537188", "#CBB279"],
padding=10,
)
)
gene_profile.add_right(
ma.plotter.Labels(
display_cells,
text_props={"color": [m[c] for c in lineage_cells]},
align="center",
padding=10,
)
)
gene_profile.add_title("Transcriptomics Profile", fontsize=16)
protein_profile = ma.SizedHeatmap(
dataset["protein_pct_matrix"],
color=dataset["protein_exp_matrix"],
cluster_data=cluster_data,
marker="*",
cmap="YlOrBr",
height=6,
width=6,
color_legend_kws={"title": "Mean Expression\n(Protein)"},
size_legend_kws={
"colors": "#de600c",
"fmt": fmt,
"title": "% expression in group",
},
)
protein_profile.group_rows(lineage_cells, order=lineage)
protein_profile.add_bottom(
ma.plotter.Labels(marker_names, color="#E36414", align="bottom", padding=10)
)
protein_profile.add_dendrogram("left", method="ward", show=False)
score = interaction["STRING Score"]
protein_profile.add_bottom(
ma.plotter.Arc(
marker_names,
interaction[["N1", "N2"]].values,
# weights=score,
colors=interaction["Type"].map({"Homo": "#864AF9", "Hetero": "#FF9BD2"}),
labels=interaction["Type"],
width=1,
legend_kws={"title": "PPI Type"},
),
size=2,
)
protein_profile.add_right(ma.plotter.Numbers(cells_count, color="#B80000"), pad=0.1)
protein_profile.add_title("Proteomics Profile", fontsize=16)
comb = gene_profile + protein_profile
comb.add_legends("top", stack_size=1, stack_by="column", align_stacks="top")
comb.render()
Total running time of the script: (0 minutes 3.806 seconds)